Morbus Gaucher ist eine autosomal rezessiv vererbte Krankheit und die am häufigsten vorkommende lysosomale Speicherkrankheit mit einer Häufigkeitsrate von etwa 1 zu 50.000 Einwohner in Zentraleuropa. Sie wird durch einen Mangel eines spezifischen Enzyms (Glucocerebrosidase) im Körper verursacht, der wiederum durch eine von beiden Elternteilen vererbte genetische Mutation verursacht wird (autosomal rezessive Vererbung). Dieser führt zur Anreicherung des Substrats Glucocerebrosid, welches in Milz, Leber, Nieren, Lungen, Gehirn und Knochenmark eingelagert wird. Jeder Krankheitsverlauf kann unterschiedlich sein, die Variationen reichen von keinerlei äußerlichen Symptomen bis hin zu schwerer Behinderung und Tod.
Die Symptome sind u.a. vergrößerte Milz und Leber, Funktionsstörung der Leber, Knochenkrankheiten oder schmerzhafte Knochendefekte, neurologische Komplikationen, Anschwellen der Lymphknoten und (gelegentlich) Gelenkdefekte, aufgeblähter Bauch, bräunliche Hautfärbung, Blutarmut, niedrige Blutplättchenzahl und gelbe Fettablagerungen auf der Lederhaut des Auges. Die am schwersten betroffenen Personen können auch für Infektionen anfälliger sein. Trotz der Tatsache, dass Morbus Gaucher aus einem Phänotyp mit unterschiedlichen Schweregraden besteht, wurde dieser, abhängig vom Vorhandensein oder Fehlen einer neurologischen Beteiligung, in drei Subtypen unterteilt.

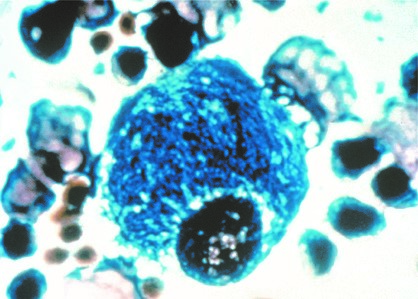
Morbus Gaucher wird als „Lipidspeicherkrankheit“ bezeichnet, bei der anormale Mengen an Lipiden, sogenannte „Glycosphingolipide“, in speziellen retikuloendothelialen Zellen gespeichert werden. In klassischen Fällen wird der Zellkern an den Rand der Zelle geschoben und die Zelle mit anormalen Lipiden gefüllt.
Morbus Gaucher Typen
Typ I, der häufigste, auch ’nicht-neuropathischer‘ Typ genannt, zeichnet sich durch die oben genannten Probleme auf. Es wird oft als Erwachsenen-Form bezeichnet, obwohl dessen Ursache schon bei der Empfängnis vorhanden ist. Das mediane Alter bei der Diagnose ist 28 Jahre und die Lebenserwartung ist leicht verkürzt. In den meisten Fällen gibt es keine wesentlichen neurologischen Symptome.
Typ II, auch akuter neuronopathischer Morbus Gaucher genannt, tritt selten auf und zeichnet sich durch schnell fortschreitende neurologische Probleme in Neugeborenen aus. Früher als infantiler Morbus Gaucher bezeichnet, zeichnet sich Typ II durch schwere neurologische Beteiligung im ersten Lebensjahr aus. Weniger als 1 in 100.000 Neugeborenen haben Typ 2. Die Prognose ist düster: wegen der schweren Beteiligung des Nervensystems wird ein betroffenes Kind in den seltensten Fällen älter als 2 Jahre.
Typ III, auch neuronopathischer Morbus Gaucher genannt, ist auch sehr selten aber im Gegensatz zu Typ II wird er durch langsam fortschreitende neurologische Symptome charakterisiert. Die Zeichen und Symptome für einen Morbus Gaucher Typ III treten später in der Kindheit auf als die Symptome beim Typ II.

Der Name der Erkrankung stammt von seinem Entdecker, Philippe Charles Ernest Gaucher (26. Juli 1854 –25. Januar 1918), einem französischen Dermatologen. 1882, als er noch Student war, entdeckte er die Krankheit an einer 32 Jahre alten Frau, die eine vergrößerte Milz hatte.